
Background
Non-small cell lung cancer (NSCLC) is the one of the most lethal cancer types worldwide1, with treatment options that have historically been limited due to factors like late-stage diagnosis. Antibody-drug conjugates (ADCs) are a promising therapeutic approach that have recently transitioned from research laboratories into patient care. Unlike conventional chemotherapy, which affects cells throughout the body, ADCs combine chemotherapy drugs with monoclonal antibodies that target specific cells, improving efficacy and safety while providing new treatment options for cancer patients.
Our study presented at this year's AACR meeting focused on two emerging ADC targets that are of particular interest for the treatment of NSCLC: TROP-2 and cMET. These biomarkers have shown early clinical success as therapeutic targets for NSCLC. To effectively implement ADCs targeting these markers in clinical practice, physicians will need standardized, efficient methods to identify which patients express sufficient levels of these markers to benefit from treatment. Patient selection is critical for therapeutic success, since ADCs are only effective when their targets are present.
However, the challenge remains in developing reliable, reproducible methods to accurately quantify biomarker expression levels across diverse patient samples. To address this challenge, our team developed and evaluated a series of machine learning models to automatically quantify biomarker expression in NSCLC samples. These models provide an automated scoring system that can assist physicians in their evaluations of when to use targeted therapies. Our approach was designed to test model generalizability – we initially trained an expression scoring model on TROP-2 data, then evaluated performance on cMET samples as a proof-of-concept.
A generalizable model for biomarker assessment could potentially eliminate the need to develop and validate separate models for each biomarker of interest, reducing development time and cost.
Methods
In this study, we developed a series of AI models for ADC biomarker evaluation. Tissue microarrays (TMAs) were generated with tumor cores from 1,142 NSCLC patients at two institutions - Charité Berlin and University Hospital Cologne. The TMAs were first immunohistochemically (IHC) stained for TROP-2 and cMET, and then digitized for AI analysis.
Our series of machine learning models performed three sequential analyses:
- Nucleus detection to identify individual cells
- Cell classification to highlight tumor cells
- Cell expression scoring to quantify membrane expression in detected tumor cells
To validate cross-marker generalizability, the model was trained using TROP-2 stained slides and evaluated on both TROP-2 and cMET. Generalization should be enhanced by the pathology foundation model (RudolfV) backbone.
Results
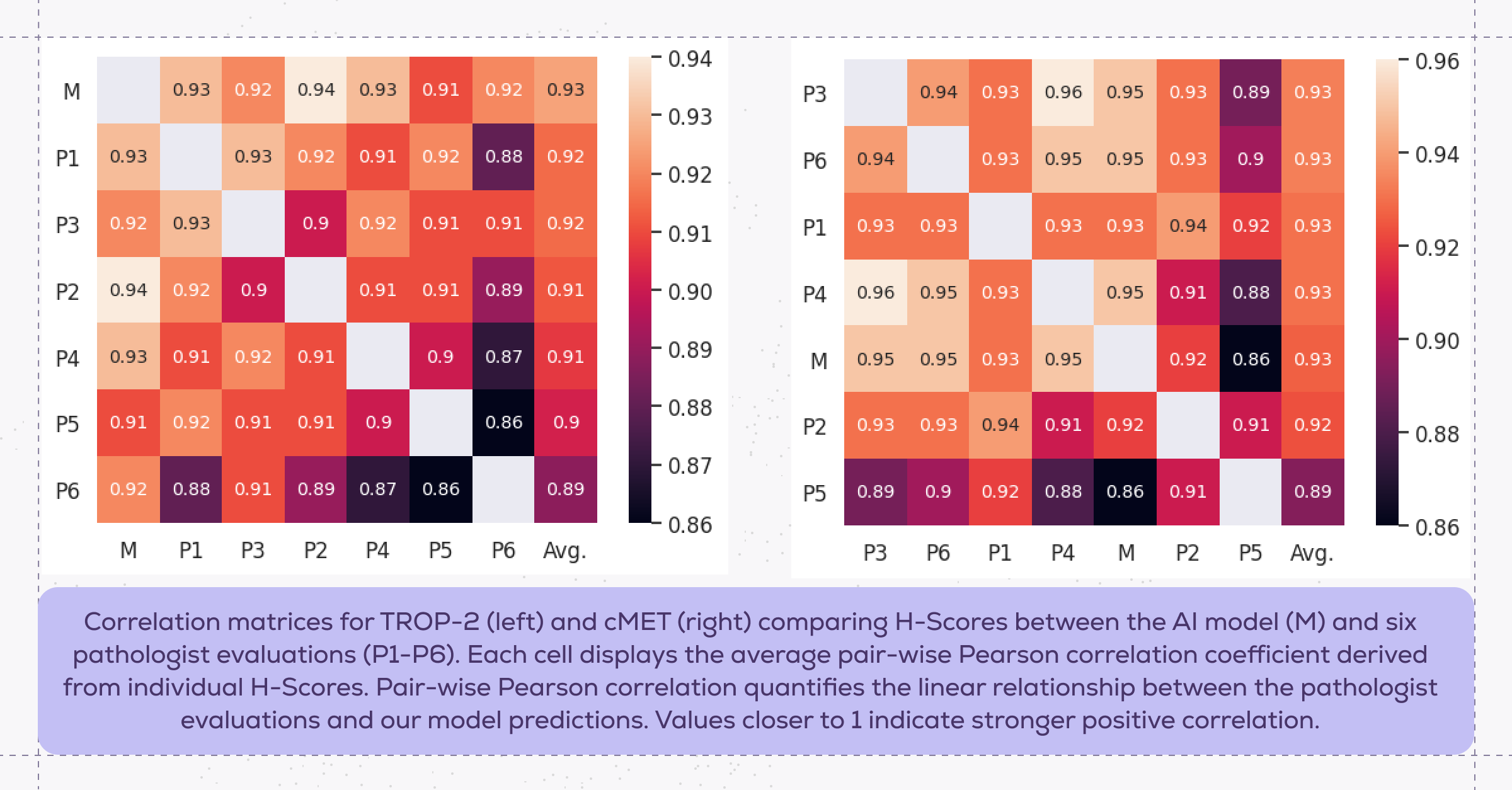
Performance of the TROP-2 expression model on TROP-2 and cMET samples was evaluated by comparing model predictions against manual H-score evaluations performed by six pathologists on a hold-out dataset of 100 slides for each marker. H-scoring is a semi-quantitative method that is calculated by multiplying the percentage of positively stained cells with the intensity of staining:
H-score = (1 × % of cells with weak intensity) + (2 × % of cells with moderate intensity) + (3 × % of cells with strong intensity)
This calculation produced H-scores ranging from 0-300, which were grouped into four expression ranges (negative=0-50, weak=50-100, moderate=100-200, strong=200-300). Higher scores indicate stronger biomarker expression, which is associated with higher likelihood of response to ADC therapy.
In evaluating the model predictions against pathologist evaluations, we found that the grouped H-scores predicted by the model demonstrated high agreement with the grouped pathologists' scores, with an average pair-wise Pearson correlation of 93% for both TROP-2 and cMET. This strong correlation indicates that our expression scoring model achieved excellent concordance with expert pathologist evaluation.
Notably, the expression scoring model, which was trained exclusively on TROP-2, generalized effectively to cMET without requiring additional fine-tuning. These results support our earlier hypothesis that using RudolfV as the basis of the model enabled this cross-marker generalizability.
Conclusion
This study demonstrates the potential of AI models to address key challenges in ADC biomarker evaluation for NSCLC. The strong alignment between our model predictions and pathologist assessments demonstrates the value of our automated scoring approach.
More importantly, our model's ability to generalize between biomarkers highlights its adaptability. This establishes the potential for automated scoring approaches to facilitate rapid expansion to additional biomarkers, streamlining treatment decision pathways for NSCLC patients.
In the future, we plan to expand our approach to investigate additional ADC-relevant biomarkers and further study the generalization capabilities of our models.
___________________________________________________
- World Health Organization. "Lung Cancer." World Health Organization Fact Sheets, published June 26, 2023. https://www.who.int/news-room/fact-sheets/detail/lung-cancer.